

DISCAPACIDAD
VISUAL

AMBLIOPÍA
Descripción
La ambliopía (ojo vago), consiste en el deterioro progresivo, mayor o menor, de la visión de un ojo. No hay lesión orgánica o esta no es proporcional a la intensidad de la disminución de la agudeza visual. Suele ser monocular.
Los siguientes factores juegan un papel fundamental:
1. Ausencia de una adecuada estimulación visual del ojo afectado.
2. Marcada dominancia de uno de los ojos.
3. Estos factores deben tener lugar a una edad temprana, desde el nacimiento hasta los siete años En esta etapa la ambliopía es reversible con el tratamiento adecuado, por lo que es importantísimo el diagnóstico precoz.
Causas
✓ La presencia de un estrabismo es el origen más frecuente; el niño fija más frecuentemente con el ojo contrario al ambliope o vago.
✓ Otra causa es la anisometropía o diferencia de graduación de un ojo a otro, que puede favorecer el retraso del desarrollo de la capacidad visual del ojo con mayor defecto, favoreciendo la ambliopía de dicho ojo.

✓ Causas oculares propias. La catarata congénita es la más frecuente.
✓ El principal factor de riesgo es la existencia de antecedentes familiares.
Diagnóstico
Ante los siguientes signos es conveniente que el bebé se someta a exploración por el especialista:
✓ Desviación ocular.
✓ Mancha blanca en la pupila.
✓ Intensa fotofobia (daño a la luz).
Es indispensable la valoración de la agudeza visual por el oftalmólogo desde los primeros meses de la vida del niño.
Pronóstico
El éxito será mejor cuanto más joven sea el niño y mejor sea la agudeza visual antes de iniciar el tratamiento.
Tratamiento
El tratamiento persigue dos objetivos:
✓ Recuperar la agudeza visual máxima.
✓ Mantenimiento de esta recuperación. Se realiza con la llamada “Penalización”, que consiste en la disminución de la agudeza visual de un ojo mediante una corrección óptica inexacta o con la utilización de “laca”.
El tratamiento debe ser:
✓ Precoz. En edades adultas la ambliopía es irreversible.
✓ El método más normalmente aceptado es la oclusión con parches sobre el ojo sano. La pauta será diferente según los casos y la vigilancia será extrema, para irlo acoplando a las necesidades de cada momento.
✓ Durante el tratamiento puede que el niño tuerza el ojo y puede disminuir la agudeza visual del ojo sano, pero ambas circunstancias son solucionables.
✓ El niño debe ser controlado periódicamente hasta los doce años, momento en que dejan de ser sensorialmente sensibles.
ANIRIDIA
Descripción
El iris es la estructura de color que se encuentra debajo de la córnea y cuyo orificio central constituye la pupila.

El color del iris da la tonalidad de nuestros ojos. Es responsable en parte de regular la cantidad de luz que debe entrar en el ojo. Realiza la función de diafragma del ojo. Presenta un músculo de disposición circular que permite modificar el tamaño de la pupila. El término aniridia hace mención a la falta del iris del ojo, pero en realidad se encuentran involucradas más estructuras. Suele afectar a ambos ojos y no es completa, hay un iris incipiente que no ha llegado a desarrollarse.
Es importante que la sociedad conozca la enfermedad para evitar el rechazo social que se da en muchas ocasiones. En los colegios hay que enseñar a los niños que su compañero es totalmente normal, explicarles porqué tienen que utilizar gafas de sol, los problemas que puede tener para la práctica de determinados deportes, el porqué de su mirada especial, etc.
Causas
Existen dos tipos de aniridia, la hereditaria y la esporádica, en la que no existen antecedentes familiares de la enfermedad, pero que una vez adquirida se convierte en hereditaria. El patrón hereditario es autosómico dominante (la transmite el portador al 50% de su descendencia). Hay una falta de desarrollo del globo ocular durante el embarazo debido a una mutación genética. Se encuentra alterado el brazo corto del par 13 del cromosoma 11 y falta de proteínas en el ADN en el gen Pax 6, responsable de la formación el ojo. Debido a esto se encuentran alteradas varias estructuras del ojo e, incluso, otros órganos del cuerpo. Las familias afectadas deberían someterse a un estudio genético.
Epidemiología
Es una enfermedad de baja incidencia; se da aproximadamente 1 caso de cada 80000-100000 nacimientos.
Síntomas
✓ El síntoma fundamental es la fotofobia (intolerancia anormal para la luz).
✓ Hay baja agudeza visual, entre un 20 – 10%, que se agrava en el caso que la aniridia se acompañe de otras lesiones oculares como catarata o glaucoma congénito.
✓ Dificultad para la visión lejana, por lo que para ver los objetos con nitidez tiene que acercarse
mucho a ellos.
✓ No distingue los detalles de un objeto a contraluz.
✓ Para leer necesita luz directa sobre el papel y que no existan sombras ni reflejos sobre cristales o espejos.
Alteraciones asociadas
✓ Nistagmus (movimientos involuntarios del ojo que impide fijar la visión).
✓ Cataratas
✓ Glaucoma.
✓ Degeneración corneal.
✓ Hipoplasia macular y del nervio óptico.
✓ Estrabismo
✓ Ambliopía: ojo “vago”, se utiliza más el ojo de mejor visión. Se trata mediante parches, etc.
✓ Luxación del cristalino.
✓ Ojo seco.
✓ Tumor de Wilms; consiste en un tumor en el riñón que se acompaña en ocasiones de retraso mental y alteraciones genitourinarias. Es importante realizar un diagnóstico precoz de la enfermedad para que no se extienda a otras partes el cuerpo y, en este caso, la supervivencia es alta.
✓ Ataxia cerebelar.
Diagnóstico
El niño recién nacido cierra los ojos cuando hay luz y se encuentra más cómodo en la penumbra. Ante la postura del niño, debe ser revisado por un oftalmólogo para determinar el grado de afección y las posibles alteraciones asociadas. Es importante realizar chequeos periódicamente para descartar posibles alteraciones renales o de otras zonas el organismo. Se debe visitar el oftalmólogo cada tres o seis meses durante los primeros años de vida y posteriormente al menos una vez al año.
Tratamiento
 En la actualidad no hay un tratamiento efectivo para la aniridia, tratándose sólo las alteraciones asociadas. La aniridia se padece desde el nacimiento, por lo que es conveniente que el niño sea consciente del problema desde el principio y de una manera progresiva. Tiene dificultad para leer, distinguir determinados objetos y para realizar determinadas actividades.
En la actualidad no hay un tratamiento efectivo para la aniridia, tratándose sólo las alteraciones asociadas. La aniridia se padece desde el nacimiento, por lo que es conveniente que el niño sea consciente del problema desde el principio y de una manera progresiva. Tiene dificultad para leer, distinguir determinados objetos y para realizar determinadas actividades.
Hay que hacerle comprender que como tiene la pupila muy grande le hace más daño la luz que a otros niños y es necesario el uso de gafas de sol y otras ayudas visuales para realizar determinadas actividades. No es conveniente dramatizar la situación y procurar que haga una vida totalmente normal, como el resto de sus compañeros y amigos. Es aconsejable la asistencia a un centro escolar donde con ayudas visuales y la cooperación del profesorado pueda realizar exactamente las mismas actividades que el resto de sus compañeros y que la integración sea total. Las lentillas cosméticas con iris artificial pigmentado alivian la fotofobia y disminuyen el nistagmus (movimientos involuntarios del globo ocular), aunque no siempre mejoran la visión. Las lentes convencionales no suelen mejorar la agudeza visual por lo que tienen que utilizar lentes de baja visión.
Las ayudas visuales que se pueden considerar:
✓ Gafas – lupa
✓ Atril.
✓ Flexo con lupa.
✓ Libros adaptados con letra grande.
✓ Programas específicos de ordenador.
ATROFIA ÓPTICA DE LEBER
Descripción
La atrofia óptica es un término clínico genérico aplicado a ciertos cuadros de palidez, avascularidad, o excavación de la papila que reflejan a posteriori la extensión del daño degenerativo subyacente. La degeneración neurológica atrófica es un proceso irreversible. La atrofia del nervio óptico es una incapacidad permanente de la vista causada por daños al nervio óptico. El nervio óptico es como un cable (mas de un millón de nervios pequeños, axones) que lleva información del ojo al cerebro para ser procesada. Cuando alguno de estos nervios han sido dañados por alguna enfermedad, el cerebro no recibe la información completa y la vista se nubla. La atrofia puede variar desde parcial, cuando algunos de los axones están dañados, hasta profunda, cuando lo están la mayoría. Puede afectar a un ojo o a los dos y también puede ser progresiva, dependiendo de la causa. Las áreas del ojo más vulnerables son las correspondientes a la zona central de la retina, la zona responsable de los detalles y el color (mácula).
La atrofia óptica hereditaria de Leber es una enfermedad desmielinizante que produce ceguera parcial, dándose en varones. Los síntomas iniciales suelen aparecer al final de la adolescencia o hacia los 20 años. Se hereda a través de la madre.

Causas
Muchas enfermedades pueden dar comienzo a la atrofia óptica:
✓ Tumores. Atrofia compresiva tumoral
✓ Falta de sangre y oxígeno. Atrofia isquémica.
✓ Trauma.
✓ Hidrocefalia.
✓ Herencia: lleva un patrón dominante, por lo que los padres con este padecimiento transmiten la enfermedad a la mitad de sus hijos. Varios equipos de científicos franceses, ingleses y alemanes han descubierto el gen OPA1 responsable de la atrofia óptica dominante de tipo I.
✓ Procesos inflamatorios: principalmente la neuritis óptica.
✓ Atrofia tóxica.
Epidemiología
La epidemiología es el estudio de la distribución y las determinantes de la frecuencia de enfermedad en el hombre.
La atrofia óptica hereditaria de Leber afecta a una entre 12000 y 50000 personas y es la neuropatía óptica hereditaria más frecuente.
Síntomas
✓ Se encuentra afectada la vista central.
✓ Puede haber deficiencias en la vista del color.
✓ Puede haber dificultad para ver contrastes.
✓ Pérdida de acuidad visual (habilidad para ver claramente), que puede variar desde vista prácticamente normal a ceguera total.
Diagnóstico
Lo diagnosticará el oftalmólogo de varias maneras:
✓ Descubriendo que la habilidad de ver claramente y la visión de color es anormal.
✓ La pupila reacciona menos de lo normal a la luz.
✓ Con el oftalmoscopio, el nervio óptico tiene un color blanquecino grisáceo.
✓ La atrofia óptica que ocurre en ambos ojos desde el nacimiento (bilateral y congénita) puede producir movimientos involuntarios en el ojo (nistagmus).
Tratamiento
Es importante detectar precozmente una atrofia óptica, para poder tratarla, detener el proceso y preservar la visión. La atrofia óptica es un signo de enfermedad crónica del nervio óptico más que un diagnóstico en sí misma; su presencia exige estudiar la causa. La resolución de ciertos procesos patológicos puede acompañarse de una recuperación visual espectacular (pueden desaparecer los defectos de agudeza visual y del campo tras eliminar la compresión del nervio óptico debida a un tumor). La identificación del gen OPA1 puede permitir el descubrimiento de nuevas terapias para tratar la enfermedad, pero por ahora lo que sí permitirá es un diagnóstico genético de la atrofia óptica dominante y ofrecer un consejo genético a las familias de riesgo.
La atrofia óptica de Leber no tiene tratamiento.
Niños
✓ Determinar la mejor posición en la que se debe colocar al niño y sus juguetes para aprovechar su vista lo mejor posible.
✓ Permite que el niño descanse entre actividad y actividad.
✓ Describa con el tacto y con palabras las actividades que se están llevando a cabo.
✓ Ambiente con buen contraste e iluminación par poder ver los objetos más claramente. Juguetes de color claro sobre fondo oscuro, etc.
✓ Use objetos reales y con los que esté familiarizado para estimular su atención visual. No cambiar nada del objeto con el que está familiarizado hasta que él pueda reconocerlo consistentemente (cuando el niño reconozca perfectamente su taza de un determinado color ya le puede cambiar el color).
✓ Cuando introduzca un objeto nuevo, procure que tenga alguna relación con otro con el que está familiarizado.
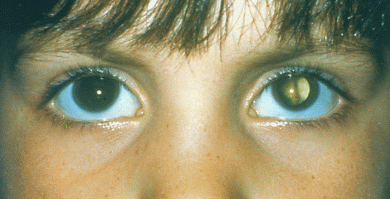
CATARATA CONGÉNITA
Definición
Las cataratas congénitas son aquellas opacidades del cristalino que se presentan en los tres primeros meses de vida. Se consideran las anormalidades oculares más comunes y suponen una causa importante de deterioro visual en la niñez.
Es la enfermedad responsable entre el 10 y el 39% de todas las cegueras ocasionadas en los niños.
Causas
Hereditarias.- Cerca de una tercera parte de las cataratas congénitas son de origen hereditario.
Causadas por infecciones intrauterinas
✓ Rubéola.- Un 15% de las mujeres en edad fértil son susceptibles de padecer la infección por el virus. La infección natural confiere inmunidad permanente. La infección fetal es probable que se de cómo exultado de la viremia de la madre con siembra del virus en la placenta. El riesgo fetal guarda relación con el momento de a gestación: 50% durante las primeras 8 semanas, 33% entre la 9ª y la 12ª semana y 10% entre la 13ª y la 24ª semana de gestación. Los defectos oculares se presentan en el 30 al 60% de los niños afectados. La catarata es el resultado de la infección antes de la 9ª semana de gestación. Puede afectar a ambos ojos y en ocasiones se observa desde el nacimiento.
Hay una opacidad central blanca, densa y una opacidad menor de la corteza circundante. Entre otras alteraciones oculares que se pueden presentar se encuentra el glaucoma. La mejor prevención es la vacunación.
✓ Herpes simple.
✓ Citomegalovirus.
✓ Toxoplasmosis.
Causadas por desórdenes metabólicos
✓ Galactosemia.- Se debe a un error innato del metabolismo de la galactosa. En presencia del azúcar de la leche se produce el depósito de una sustancia (galactitol) en el cristalino produciendo su opacificación. No suele manifestarse al nacer y se hace aparente en los primeros meses de la vida. Se presenta en los niños afectados por la enfermedad y son alimentados con productos lácteos que contiene lactosa (glucosa + galactosa). Además de la catarata se puede presentar retardo mental, inhibición del crecimiento y disfunción hepática. Con el diagnóstico temprano y la aplicación rápida de la terapia por la eliminación de la galactosa de la dieta se puede regresar la catarata y mejorar las otras señales de la enfermedad.
✓ Déficit de galactoquinasa.- Su carencia conduce a la acumulación de galactosa, ocurriendo lo mismo que en la galactosemia. A excepción de la catarata tienen una salud normal. El tratamiento consiste en la eliminación de la galactosa de la dieta.
✓ Hipoglicemia neonatal.- Se da en el 20% de los recién nacidos, siendo más frecuente en los prematuros. Generalmente se presenta a los dos o tres años de vida y muchos niños no tienen
incapacidad visual ninguna.
✓ Hipoparatiroidismo.
Asociada a otras anomalías oculares
✓ Microftalmia.
✓ Glaucoma congénito.
Por ingestión de medicamentos por parte de la madre
✓ Corticoides.
✓ Sulfonamidas.
Radiación
✓ Rayos X durante el primer trimestre del embarazo.
Malnutrición materna
✓ Falta de vitamina A, B1, C, D.
✓ Falta de ácido fólico.
Asociados a síndromes
✓ Sin causa conocida
✓ Idiopáticas. Afecta a un tercio de los niños y no tienen ninguna otra enfermedad asociada. Muchos de estos casos son por mutaciones nuevas y la naturaleza familiar de la catarata se descubrirá en las siguientes generaciones.
Epidemiología
La presencia de catarata infantil se ha estimado entre 1 a 15/10000 niños. La incidencia de catarata congénita bilateral en los países industrializado es de 1 – 3/10000 nacimiento, siendo probablemente mayor en los países subdesarrollados, debido a diversos factores etiológicos como la rubéola.
La prevalencia de ceguera (agudeza visual con corrección menor a 0.05) por catarata infantil puede estar alrededor de 0.4/10000 niños en los países industrializados.
Síntomas
En los adultos hay disminución de la agudeza visual, pero en los niños suele ser unilateral y asintomática, por lo que se da un retraso en el diagnóstico.
Las manifestaciones más importantes son:
✓ Leucocoria: es un reflejo blanco que puede ser visto por los padres o por el pediatra.
✓ Nistagmus: espasmo de los músculos motores del globo ocular produciendo movimientos involuntarios de éste en varios sentidos: horizontal, vertical, oscilatorio, rotatorio o mixto. Su presencia sugiere mala agudeza visual.
✓ Estrabismo: debido a la agudeza visual disminuida.
✓ Fotofobia (intolerancia anormal para la luz).
Diagnóstico
En la evaluación del niño con catarata hay que tener en consideración tres aspectos:
Examen oftalmológico
✓ Inspección
✓ Exploración de la agudeza visual: permitirá determinar el momento de la operación.
✓ Determinación de la refracción: en niños mayores de tres años.
✓ Dilatación del ojo y exploración con el oftalmoscopio.
✓ Etc. (estudio de la motilidad ocular, estudio del nistagmus, estudiar la existencia de otras enfermedades asociadas).
Estudio del paciente
✓ Valoración completa del niño que debe realizar su pediatra, prestando especial atención a la presencia de enfermedades asociadas y eventual retraso mental, frecuente en muchos de estos niños.
Estudio de los padres
✓ Búsqueda de la causa de la catarata
o Hereditarias: los padres y los hermanos deberían ser examinados.
o Infecciones intrauterinas maternas.
o Ingestión de fármacos durante el embarazo.
o Radiaciones durante el embarazo.
✓ Postura
* Motivación de los padres y su comprensión y buena voluntad para participar en el cuidado postoperatorio de su hijo que con frecuencia es complicado.
Pronóstico
El pronóstico de los ojos operados dependerá, en gran medida, del momento de la operación y de la rehabilitación visual postoperatoria: tratamiento de la ambliopía (oscurecimiento de la visión por sensibilidad imperfecta de la retina).
Tratamiento
El único tratamiento posible es el quirúrgico. La cirugía ocular en la edad pediátrica tiene unas consideraciones diferentes que en el adulto. Primero, los niños tienen una larga expectativa de vida y, por otro lado, hay una respuesta diferente a la agresión quirúrgica, teniendo una posibilidad de recuperación mayor. Hay que tener en cuenta que los niños representan una población de mayor riesgo durante la anestesia.
 En el caso de catarata congénita total, la cirugía debe realizarse de una manera precoz; si la catarata es parcial y es monocular hay que hacer un tratamiento previo refractario y oclusivo y si la agudeza visual mejora esperar. En el caso de que empeore hay que operar e instaurar tratamiento rehabilitador. En el caso de bilaterales parciales el tratamiento es refractario, oclusivo del ojo mejor y si mejora la agudeza visual realizar
En el caso de catarata congénita total, la cirugía debe realizarse de una manera precoz; si la catarata es parcial y es monocular hay que hacer un tratamiento previo refractario y oclusivo y si la agudeza visual mejora esperar. En el caso de que empeore hay que operar e instaurar tratamiento rehabilitador. En el caso de bilaterales parciales el tratamiento es refractario, oclusivo del ojo mejor y si mejora la agudeza visual realizar
controles periódicos y esperar; si no mejora, operar el ojo peor y tratamiento rehabilitador de este ojo, con controles del ojo mejor. Si la agudeza visual disminuye hay que realizar cirugía inmediata de dicho ojo y tratamiento rehabilitador. Una vez decidida la intervención quirúrgica, hay que realizarla cuanto antes, pues los resultados obtenidos dependerán directamente de la precocidad de la cirugía.
Las dos técnicas más empleadas son:
1. Lensectomía (extracción completa del cristalino).
2. Extracción del cristalino e implante de lente intraocular. Normalmente no se utiliza en menores de 1 año a no ser que exista muy poca colaboración familiar en donde la adaptación de lente de contacto y el tratamiento de la ambliopía van a ser imposibles. Los mejores candidatos para esta técnica son las formas bilaterales que presentan sólo catarata y ojo bien formado. Algunos estudios indican que el implante de una lente intraocular del tamaño adecuado al ojo del niño pequeño puede tener un efecto beneficioso sobre el crecimiento del ojo, viéndose un crecimiento muy similar al del ojo normal.
La decisión tiene que ser individualizada y en función del grado de opacificación, enfermedad ocular asociada, edad y grado de colaboración del paciente y su familia.
Medidas preventivas
Es fundamental un diagnóstico precoz.
Rehabilitación
La rehabilitación postoperatoria tiene tres aspectos:
1. Corrección de la ametropía residual. Siempre que sea posible se realizará con lentes de contacto.
2. Tratamiento de la ambliopía y conservación de la agudeza visual.
3. Corrección de las anomalías asociadas: glaucoma, estrabismo, alteraciones corneales, etc.
CEGUERA
Definición
Es la pérdida de la visión normal o que se pueda corregir.
Consideraciones generales
La ceguera puede ser parcial, con pérdida de solamente una parte de la visión o también total, en cuyo caso la persona no tiene ninguna percepción de la luz. Las personas con una visión inferior a 20/200 se consideran ciegos en términos legales. La ceguera tiene muchas causas, sin embargo, la incidencia de ceguera verdadera es aún baja en los Estados Unidos en donde la mayoría de los casos se deben a accidentes, diabetes, glaucoma y degeneración macular. A nivel mundial la causa más significativa es la deficiencia de vitamina A.
Causas comunes
∙ Trauma ocular accidental como quemaduras químicas o lesiones infringidas por las cuerdas del salto Bungy, anzuelos de pesca, pelotas de raqueta, juegos pirotécnicos y objetos similares
∙ Diabetes foto
 ∙ Glaucoma
∙ Glaucoma
∙ Degeneración macular
∙ Oclusiones vasculares
∙ Ambliopía
Otras causas menos comunes son:
∙ Neuritis óptica
∙ Período de poscirugía ocular
∙ Apoplejía
∙ Deficiencia de vitamina A
∙ Enfermedad de Tay Sachs
∙ Retinitis pigmentosa
∙ Retinoblastoma
∙ Intoxicación por plomo
∙ Glioma óptico
∙ Tracoma (conjuntivitis por Clamidia)
 Otras causas menos frecuentes son:
Otras causas menos frecuentes son:
∙ Síndrome de Jansky Bielschowsky
∙ Enfermedad de Krabbe
∙ Fibroplasia retrolental
∙ Acromatopsia
∙ Enfermedad de Albers Schonberg (osteopetrosis)
∙ Degeneración cerebral difusa de Alpers
∙ Anoftalmos
∙ Batten Mayou
∙ Síndrome de Cockayne
∙ Criptoftalmía
∙ Oftalmia gonocócica
∙ Enfermedad de Kufs
∙ Amaurosis congénita de Leber
∙ Enfermedad de Niemann Pick
∙ Enfermedad de Norrie
∙ Oncocerciasis (ceguera de los ríos)
∙ Síndrome de Refsum
∙ Enfermedad de Scholz
∙ Trisomía 13
∙ Vogt Spielmeyer
DISTROFIA CORNEAL
Descripción
Las distrofias corneales son enfermedades raras que afectan a la capa clara más externa del globo ocular. El término distrofia se refiere a una alteración del volumen y peso de un órgano. Son afecciones bilaterales que al principio afectan a la zona central y excepcionalmente a la zona marginal.
 Causas
Causas
Las distrofias corneales se deben a una alteración del metabolismo del ojo y su periferia, más frecuentes en los varones y en la adolescencia.
Síntomas
Existe una opacidad en la córnea que provoca alteración de la agudeza visual. Se forman pequeñas áreas de opacidades superficiales debidas a la aparición de fibrosis entre las capas de la córnea que avanzan según evoluciona la enfermedad. Son prácticamente asintomáticas y no asocian queratitis (es una enfermedad en la que se produce un aumento del desarrollo y engrosamiento del epitelio de la capa córnea de la piel).
Son de progresión muy lenta, pero sobre los 40 años la visión se puede ver seriamente comprometida, por lo que se recomienda practicar una queratoplastia. (El término Queratoplastia se refiere al transplante o injerto corneal, es decir, el tejido corneal enfermo es sustituido por un tejido donante). Dentro de las distrofias corneales existe una forma especial denominada enfermedad de Fuchs, más frecuente en las mujeres mayores,
en la que hay atrofia con edema, formación de vesículas y opacidades puntiformes en la córnea. Los enfermos ven halos corneales por la mañana que van despareciendo a lo largo del día. En las últimas etapas la córnea se vuelve totalmente opaca e insensible.
Pronóstico
Debido a que el proceso es muy lento, aunque su curso hacia la ceguera sea irreversible, los pacientes fallecen antes de que ésta se produzca.
Tratamiento
Puede ser necesario practicar una queratoplastia (trasplante de córnea).
DISTROFIA RETINAL
Descripción
Las distrofias retinianas son una serie de enfermedades hereditarias de la retina que conducen a la degeneración de los fotorreceptores.
Los fotorreceptores son unas células dispersas por la retina especializadas en recibir el estímulo luminoso. Se conocen dos tipos de fotorreceptores, los conos y los bastones (por su forma). Los conos son características de la visión con buena iluminación y responsables de la agudeza visual y de la percepción del color. Hay tres tipos de conos, cada uno sensible a los tres colores primarios: rojo, verde y azul. Los bastones dan lugar a la visión en blanco y negro y con poca capacidad para diferenciar detalles, en condiciones de baja iluminación ambiental.
Una forma especial de este grupo de enfermedades es la Amaurosis Congénita de Leber (LCA), en la que la pérdida de visión suele manifestarse a los pocos meses de vida.
Causas
La Amaurosis Congénita de Leber es una enfermedad hereditaria que sigue un patrón autosómico recesivo.
Síntomas
En la Amaurosis de Leber nos encontramos con pérdida de visión que se acompaña a menudo de nistagmus (espasmo de los músculos motores del globo ocular que produce movimientos involuntarios de éste) y reflejo pupilar anómalo. Frecuentemente se observa pigmentación retiniana y signos de atrofia.
Tratamiento
Se ha visto la mutación genética responsable del 2 al 16% de casos de LCA: gen RPE65, en el cromosoma 1p31 y se está investigando en este campo con la inyección subretinal de AAVRPE65, que se cree es capaz de corregir la enfermedad en humanos, pero es necesario estudios a más largo plazo en relación con seguridad y eficacia.

Ya se están probando en Estados Unidos y en Alemania los nuevosimplantes electrónicos para recuperar la visión en personas que sufren de distrofia de retina
ESTRABISMO
Definición
Es una condición de desviación del alineamiento anormal de un ojo con respecto al otro.
Causas, incidencia y factores de riesgo
El estrabismo es producto de falta de coordinación entre los ojos que hace que los ojos apunten en direcciones diferentes y sean incapaces de enfocar un mismo punto simultáneamente. La mayoría de los casos de estrabismo de los niños tienen una causa desconocida; más de la mitad se presentan al nacimiento o poco después de nacer (estrabismo congénito).
 En los niños, cuando los dos ojos no logran enfocarse en la misma imagen, el cerebro puede aprender a ignorar la información que entra por un ojo y si esto se deja continuar, el ojo que el cerebro ignora nunca verá bien.
En los niños, cuando los dos ojos no logran enfocarse en la misma imagen, el cerebro puede aprender a ignorar la información que entra por un ojo y si esto se deja continuar, el ojo que el cerebro ignora nunca verá bien.
Esta pérdida de la visión se llama ambliopía y frecuentemente está asociada con el estrabismo.
Algunos otros trastornos asociados con el estrabismo en los niños son:
∙ Retinopatía de la prematurez
∙ Retinoblastoma
∙ Lesión cerebral traumática
∙ Hemangioma cerca del ojo durante la infancia
∙ Síndrome de Apert
∙ Síndrome de Noonan
∙ Síndrome de Prader-Willi
∙ Trisomía 18
∙ Rubéola congénita
∙ Síndrome de incontinencia pigmentaria
∙ Parálisis cerebral
Los antecedentes familiares de la enfermedad son un factor de riesgo. La hipermetropía puede ser un factor contribuyente, así como cualquier otra enfermedad que produzca pérdida de la visión puede tener al estrabismo dentro de sus complicaciones.
Síntomas
∙ Ojos que parecen bizcos
∙ Ojos que no se alinean en la misma dirección
∙ Movimientos descoordinados del ojo
∙ Visión doble
∙ Visión sólo en un ojo con pérdida de percepción de profundidad

Signos y exámenes
El examen físico consiste en una revisión detallada de los ojos, durante la cual se le pide al paciente que mire a través de una serie de prismas para determinar la magnitud de la divergencia ocular. Los músculos del ojo se examinan para determinar la fuerza de los músculos extraoculares.
Los exámenes son:
∙ Examen oftálmico estándar
∙ Agudeza visual
∙ Examen de la retina
∙ Examen neurológico
Tratamiento
Inicialmente, se intentan estrategias para fortalecer los músculos debilitados y realinear así los ojos. Es posible que se prescriban gafas y ejercicios para el músculo ocular. En caso de presentarse ambliopía, se puede colocar un parche en el ojo preferido para forzar al niño a utilizar el ojo amblíope. Si las técnicas de fortalecimiento no tienen éxito, es posible que sea necesario recurrir a la cirugía para realinear los ojos.
Pronóstico
El defecto puede corregirse con un diagnóstico temprano, pero si se retrasa el tratamiento, la pérdida de visión en un ojo puede ser permanente.
Complicaciones
∙ Pérdida de visión en un ojo secundaria a ambliopía
∙ Vergüenza por la apariencia facial al usar el parche en el ojo
También se debe llamar en caso de presentarse problemas académicos que posiblemente podrían estar relacionados con la incapacidad del niño para ver el pizarrón o leer los materiales de estudio.
GLAUCOMA CONGÉNITO
Descripción
Bajo el término de Glaucoma Congénito se consideran una serie de enfermedades, la mayoría de origen hereditario, que se caracterizan por existir una anomalía ocular en el nacimiento responsable de un aumento de la Presión Intraocular (PIO).
El más frecuente de los Glaucomas Congénitos es el Glaucoma Congénito Primario (50%), pero es, de todas maneras, una enfermedad excepcional. Se presenta en los tres primeros años de vida. Hay un desarrollo defectuoso de las vías de salida del humor acuoso.

En las primeras semanas o meses de la vida se va a presentar lagrimeo y el niño no es capaz de mantener los ojos abiertos cuando hay luz (fotofobia). La córnea pierde transparencia y se ve blanquecina. Simultáneamente, el ojo, como consecuencia del aumento de presión en su interior, va aumentando de tamaño. Hay que tener especial cuidado con los niños que tienen los ojos mucho más grandes que el resto de los niños de su misma edad, especialmente si les molesta mucho la luz.
Causas
Es una enfermedad hereditaria que sigue un patrón autosómico recesivo de baja penetración.
Las anomalías de uno o más genes son bastante frecuentes, sobre todo de los recesivos. Se considera que un gen anormal es recesivo cuando necesita de su homólogo también anormal para desarrollar la enfermedad, cada individuo tiene de seis a ocho genes recesivos anormales, que provocan un funcionamiento anormal de las células sólo si existen dos similares, probabilidad escasa en la población general (aumenta en niños de padres con parentesco cercano y en aquellos grupos cerrados que se casan entre sí).
Cada gen controla la producción de una proteína, por lo que un gen anormal produce una proteína anormal o una cantidad anormal de la misma, lo que puede causar un funcionamiento anormal de la célula y, en definitiva, de las funciones corporales.
Si cada uno de los progenitores tiene un gen anormal y otro normal, no padece la enfermedad, pero pueden transmitir el gen anormal a su descendencia (portadores). Cada hijo tiene un 25% de probabilidades de heredar dos genes anormales (desarrolla la enfermedad), un 25% de heredar dos genes normales y un 50% de heredar un gen anormal (no padecen la enfermedad pero son portadores como los padres). Con frecuencia las causas de los rasgos genéticos anormales se debe más a nuevas mutaciones genéticas que a la herencia de los padres.
Epidemiología
El Glaucoma Congénito Primario es una enfermedad muy poco frecuente, dándose en 1 de cada 10000 recién nacidos. El 65% de los afectados son varones.
Síntomas
Se suelen afectar los dos ojos en el 75% de las ocasiones, aunque el grado de gravedad en ambos habitualmente es diferente. Hay tres manifestaciones clásicas y la presencia de cualquiera de ellas hace sugerir la sospecha de glaucoma de un bebé o niño:
✓ Lagrimeo excesivo (epífora).
✓ Rehuir de la luz (fotofobia), se esconde en los brazos de la madre. Se suele manifestar después de varios meses de vida y se acompaña generalmente de una constricción involuntaria de los párpados
(blefaroespasmo) que puede ser otra manifestación de la fotofobia.
✓ Ojos grandes. El aumento de la Presión Intraocular (PIO) provoca un agrandamiento del globo ocular y, por tanto, de la córnea.
Diagnóstico
El diagnóstico generalmente lo realizan los padres al observar los síntomas característicos que son la turbidez o edema de la córnea (pierde la transparencia y está blanca), el lagrimeo excesivo y la fotofobia.
Mediante el examen físico se comprueba:
✓ Diámetro de la córnea. foto
 ✓ Inflamación de la córnea. Con frecuencia es el primer signo observado por los padres.
✓ Inflamación de la córnea. Con frecuencia es el primer signo observado por los padres.
✓ El iris suele ser normal.
✓ Estudio del campo visual.
✓ Agudeza visual.
Se necesita hacer un diagnóstico que la diferencie (diagnóstico diferencial) de otras alteraciones:
✓ Lagrimeo excesivo: puede ser debido a una obstrucción del conducto nasolagrimal (conducto que
comunica el lagrimal con la nariz). El saco lagrimal suele tener una secreción purulenta.
✓ Alteraciones de la córnea:
o Córnea grande congénita sin glaucoma o globo ocular grande debido a una miopía alta.
o Opacificación de la córnea en la lactancia asociada a varias alteraciones:
o Anomalías del desarrollo.
o Inflamación debida a trauma obstétrico.
o Inflamación intrautérina (sífilis congénita y rubeola).
o Errores congénitos del metabolismo (mucopolisacaridosis, etc.).
✓ Otros glaucomas de la infancia.
Tratamiento
El tratamiento es quirúrgico y el realizarlo a tiempo es fundamental para preservar la visión. En ocasiones, es necesario realizar varias intervenciones hasta que se consigue el control de la tensión. Los niños deben ser controlados indefinidamente, al principio varias veces al año y luego una vez al año por lo menos durante toda su vida.
MIOPÍA PATOLÓGICA
Descripción
La miopía se considera un estado de exceso de potencia del sistema óptico del ojo, con relación a su longitud, bien porque el ojo sea demasiado largo o bien debido a que la capacidad de hacer converger la luz que tienen los medios ópticos oculares (córnea, humor acuoso, cristalino y humor vítreo) sea excesiva. La luz proveniente de lejos no va a parar a su lugar, en el fondo del ojo, sino por delante de donde tiene que ir. No se es miope para todas las distancias de visión, se es miope a partir de una distancia del ojo. La palabra miopía proviene del griego, que significa “ojos cerrados”, debido a que la gente con esta condición cierra sus párpados para ver mejor a la distancia. La miopía recibe el apelativo de “vista corta”, porque permite ver bien a distancias cortas a costa de ver mal de lejos.

En el caso de la miopía congénita, el fallo en la estructura ha aparecido antes de que el ojo recibiese los rayos del luz del mundo exterior. Es un error orgánico sin ninguna finalidad funcional, aparecido en el principio del desarrollo y que debe ser tratado cuanto antes. La miopía patológica es un estado de crecimiento anormal del ojo, asociado a cambios degenerativos en su estructura. Puede aumentar en la vida adulta y el factor genético es determinante. Hay muy poca mejoría en la agudeza visual a pesar del uso de gafas graduadas. Puede progresar rápidamente ocasionando que la retina se desgarre y desprenda.
Causas
Existe una predisposición genética; si nuestros padres o abuelos han sido miopes, nuestra predisposición a serlo será mucho mayor, pero si nos desenvolvemos en un ambiente de espacios abiertos y largas distancias, no llegaremos a desarrollar miopía.
Cuanto más joven sea una persona, más influencia sobre su sistema visual ejercerán las demandas en visión próxima, por lo que en la mayoría de los casos la miopía suele aparecer en aquel periodo de escolaridad correspondiente a un mayor uso de la visión de cerca. La miopía es la adaptación típica del sistema visual a la visión de cerca, pues el ojo está preparado para una actividad esencialmente a distancia, con la posibilidad de adaptarse, por la acomodación, a la visión próxima.
Síntomas
El primer síntoma de un ojo miope es mala visión lejana. Un miope ve mal de lejos, pero ve bien de cerca. No obstante, resulta evidente que si una persona es miope de muchas dioptrías, para ver bien de cerca tendría que acercarse mucho algo que quisiera ver bien, lo cual resulta bastante cansado e incómodo.
Diagnóstico
Normalmente la aparición del defecto visual en el niño, asociado a un comportamiento característico, lleva a los padres a consultar con el oftalmólogo, el cual, con sistemas de medida de alcance visual u optometrías y distintas correcciones con lentes convergentes o divergentes, determinará el grado de miopía y el tratamiento corrector más adecuado.
Pronóstico
 El pronóstico de la miopía depende de la edad del paciente. Si aparece antes de los 4 años debe observarse como potencialmente grave. Por encima de esta edad o con más de 8 ó 10 años, la miopía ligera hasta 6 dioptrías no es alarmante y si se pasan los 21 años sin progresión marcada probablemente el proceso se estacione y el pronóstico es bueno. En los grados intensos el pronóstico se basa en el aspecto del fondo del ojo y en la agudeza visual después de la corrección. En cualquier caso, siempre debe considerarse la posibilidad de un desprendimiento de retina o una hemorragia súbita.
El pronóstico de la miopía depende de la edad del paciente. Si aparece antes de los 4 años debe observarse como potencialmente grave. Por encima de esta edad o con más de 8 ó 10 años, la miopía ligera hasta 6 dioptrías no es alarmante y si se pasan los 21 años sin progresión marcada probablemente el proceso se estacione y el pronóstico es bueno. En los grados intensos el pronóstico se basa en el aspecto del fondo del ojo y en la agudeza visual después de la corrección. En cualquier caso, siempre debe considerarse la posibilidad de un desprendimiento de retina o una hemorragia súbita.
Si se producen atrofias en la retina aparecen escotomas o pérdidas absolutas de visión en determinadas zonas del campo visual, aparecen moscas y opacidades flotantes muy molestas y, si el fenómeno progresa, el paciente no utiliza sus ojos con comodidad en ningún momento hasta que no queda visión útil o hasta que se produce una complicación repentina como un desprendimiento de retina.
Tratamiento
La neutralización óptica de la miopía se realiza con lentes negativas o divergentes. Se toma como valor de la miopía el de la lente que la compensa, anotándose su potencial en dioptrías, con un signo (-) delante, indicativo de la lente divergente. Las lentes divergentes o negativas aumentan su potencia efectiva si se acercan al ojo, por lo que es conveniente utilizar monturas que acerquen las lentes lo más cerca posible de los ojos.
Debe mantenerse un buen estado general, con mucha vida al aire libre, ejercicio y alimentación completa, especialmente proteínas. También es importante una buena higiene visual, durante el trabajo de cerca hay que procurar una correcta iluminación, una postura fácil, vigilar el tipo de letra y evitar la fatiga ocular excesiva.
La cirugía para la corrección de miopía se aplica con mucha frecuencia en los últimos tiempos debido a que es cada vez mas segura y predecible. Permite a la persona liberarse de sus anteojos o lentes de contacto y realizar sus actividades diarias y deportes favoritos sin preocuparse por su visión.
El tipo de cirugía a realizar depende de la magnitud de su miopía. En la actualidad la mayoría de casos pueden ser corregidos mediante un procedimiento conocido como Keratomileusis In Situ Asistida por Laser (LASIK). Para miopías mayores de 10 dioptrías se prefiere el Implante de Lente Intraocular Faquico o Lente de Contacto Implantable (ICL). Las personas mayores de 40 años de edad deben consultar el procedimiento con el oftalmólogo ya que por efecto de la presbicia pueden no estar satisfechos con su visión de cerca.
Medidas preventivas
Es importante tomar ciertas precauciones, especialmente en el caso de padres o abuelos miopes.
La predisposición genética puede ser tan grande que la evolución en el desarrollo de las estructuras oculares, frente a los estímulos normales del medio, sea un proceso de difícil control. El seguimiento de las normas de higiene visual y el tratamiento optométrico adecuado, podrán mejorar su evolución.

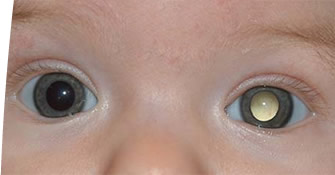
RETINOBLASTOMA
Definición
Se trata de un tumor maligno de la retina que afecta, generalmente, a niños menores de 6 años. La retina es el tejido nervioso que reviste la parte posterior de los ojos y es el encargado de detectar la luz y forma las imágenes. El tumor se puede situar en uno o en ambos ojos y normalmente no se extiende a otros tejidos o partes del cuerpo.
Causas
 Se produce una mutación en el gen RB1 (en el cromosoma 13) en una célula de la retina en crecimiento, por lo que crece sin control y se convierte en cáncer. En algunas ocasiones se desarrolla en un niño sin antecedentes familiares, aunque en otras ocasiones la mutación se encuentra presente en varios miembros de la familia: en este caso hay un 50% de posibilidades de que los hijos de la persona afectada tengan también la mutación y alto riesgo de desarrollar un retinoblastoma. Se necesitan dos mutaciones para destruir el gen RB1 y causar el crecimiento descontrolado de células. Cuando el Retinoblastoma es hereditario, la primera mutación se hereda de uno de los padres y la segunda se produce durante el desarrollo de la retina. Cuando el Retinoblastoma es esporádico (60% de los casos), ambas mutaciones se producen durante el desarrollo de la retina. El gen RB1 es un gen autosómico dominante, por lo que ambos sexos se ven igual afectados y en cada embarazo existe un 50% de posibilidades de que uno de los padres transmite el gen a su hijo. Cuando el hijo hereda el gen, hay entre un 75 y un 90% de posibilidades de que se produzca una segunda mutación; es posible que un niño que hereda la mutación no experimente la segunda y nunca desarrolle el retinoblastoma (pero sí puede transmitir el gen a sus descendientes, de forma que sus hijos pueden sufrir la enfermedad). Si uno de los progenitores tiene un Retinoblastoma unilateral, entre el 7% al 15% de sus descendientes tendrán Retinoblastoma (en el 80% aproximadamente de los casos, será bilateral). La mayoría de los Retinoblastomas que se dan en un solo ojo no son hereditarios y si se da en ambos ojos, siempre es hereditario.
Se produce una mutación en el gen RB1 (en el cromosoma 13) en una célula de la retina en crecimiento, por lo que crece sin control y se convierte en cáncer. En algunas ocasiones se desarrolla en un niño sin antecedentes familiares, aunque en otras ocasiones la mutación se encuentra presente en varios miembros de la familia: en este caso hay un 50% de posibilidades de que los hijos de la persona afectada tengan también la mutación y alto riesgo de desarrollar un retinoblastoma. Se necesitan dos mutaciones para destruir el gen RB1 y causar el crecimiento descontrolado de células. Cuando el Retinoblastoma es hereditario, la primera mutación se hereda de uno de los padres y la segunda se produce durante el desarrollo de la retina. Cuando el Retinoblastoma es esporádico (60% de los casos), ambas mutaciones se producen durante el desarrollo de la retina. El gen RB1 es un gen autosómico dominante, por lo que ambos sexos se ven igual afectados y en cada embarazo existe un 50% de posibilidades de que uno de los padres transmite el gen a su hijo. Cuando el hijo hereda el gen, hay entre un 75 y un 90% de posibilidades de que se produzca una segunda mutación; es posible que un niño que hereda la mutación no experimente la segunda y nunca desarrolle el retinoblastoma (pero sí puede transmitir el gen a sus descendientes, de forma que sus hijos pueden sufrir la enfermedad). Si uno de los progenitores tiene un Retinoblastoma unilateral, entre el 7% al 15% de sus descendientes tendrán Retinoblastoma (en el 80% aproximadamente de los casos, será bilateral). La mayoría de los Retinoblastomas que se dan en un solo ojo no son hereditarios y si se da en ambos ojos, siempre es hereditario.
Epidemiología
Poco frecuente, excepto en familias portadoras de la mutación del gen RB.
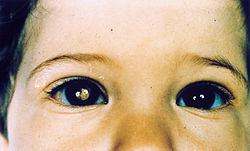 Síntomas. Anatomía del ojo
Síntomas. Anatomía del ojo
✓ Brillo blanco en el ojo.
✓ Manchas blancas en la pupila.
✓ Estrabismo: desviación de los ojos
✓ Enrojecimiento y dolor en el ojo.
✓ Visión deficiente.
✓ Iris de color diferente en cada ojo.
Existe una clasificación de la enfermedad desarrollada por dos médicos:
Clasificación de Reese-Ellsworth:
Grupo I.- Implica uno o más tumores cuyo tamaño es inferior a 4 diámetros discales y están ubicados en el
Ecuador (línea imaginaria que divide el ojo en dos partes iguales) o detrás del mismo.
Grupo II.- Implica uno o más tumores cuyo tamaño oscila entre 4 y 10 diámetros discales y están ubicados en el Ecuador o detrás del mismo.
Grupo III.- Abarca toda lesión que se encuentre frente al Ecuador o todo tumor cuyo tamaño supere los 10 diámetros discales.
Grupo IV.- Implica tumores múltiples cuyos tamaños superan los 10 diámetros discales en algunos o en todos los casos, o toda lesión que se extienda más allá de la parte posterior del ojo.
Grupo V.- Implica tumores muy grandes que abarcan más de la mitad de la retina y que se han propagado a otros partes del cuerpo
Diagnóstico
✓ Examen del ojo con dilatación de la pupila.
✓ Tomografía Computerizada (TC) de la cabeza para ver el tamaño del tumor y su posible diseminación. El TC utiliza una combinación de rayos x y tecnología computerizada para obtener imágenes de cortes transversales del cuerpo.
✓ Ultrasonido del ojo.
Si existen antecedentes familiares se pueden realizar exámenes oculares en muchas de las etapas del desarrollo del niño para determinar la presencia de un tumor. Una vez que se ha detectado el retinoblastoma, es necesario realizar otras pruebas para determinar su tamaño y comprobar si se ha diseminado a otras partes del cuerpo.
Pronóstico
El pronóstico depende, en gran medida de lo siguiente:
✓ El grado de avance de la enfermedad.
✓ Tamaño y ubicación del tumor.
✓ Presencia o ausencia de metástasis.
✓ Respuesta del tumor a la terapia.
✓ Edad y estado general del niño.
✓ Tolerancia del niño a determinados medicamentos, procedimientos o terapias.
✓ Nuevos acontecimientos en el tratamiento.
Si el cáncer no se ha diseminado fuera del ojo, casi todos los pacientes se pueden curar. En caso contrario, la probabilidad de curación es mucho más baja y depende de los órganos afectados. Más del 90% de los niños se pueden curar con la detección precoz y extirpando el ojo afectado. Los niños con retinoblastoma hereditariopueden correr el riesgo de desarrollar un tumor cerebral durante el tratamiento del tumor en los ojos (retinoblastoma trilateral), pero es un trastorno muy raro. Ciertos niños con retinoblastoma pueden desarrollar otros cánceres “no oculares”; los que tienen más riesgo son:
✓ Niños con Retinoblastoma en ambos ojos.
✓ Niños con historia familiar de Retinoblastoma.
✓ Niños con historia de múltiples tumores en su ojo.
✓ Niños que son diagnosticados de Retinoblastoma cuando tienen menos de un año.
✓ Niños que fueron tratados con irradiación cuando eran menores de un año.
Es esencial un seguimiento continuo en un niño que se le ha diagnosticado un retinoblastoma.
En resumen, con el tratamiento correcto y en manos de un oftalmólogo experimentado y de un seguimiento continuo para descartar otros cánceres, el paciente con Retinoblastoma puede tener una vida larga, completa y feliz como cualquier otra persona.

Tratamiento
El tratamiento específico lo determinará su médico basándose en:
✓ Edad, estado general y su historia médica.
✓ Grado de avance de la enfermedad.
✓ Tolerancia del niño a determinados medicamentos, procedimientos o terapias.
✓ Sus expectativas para la evolución de la enfermedad.
✓ Su opinión o preferencia.
Hay que conocer en qué etapa se encuentra la enfermedad:
✓ Retinoblastoma intraocular: el cáncer se encuentra en uno o en ambos ojos, pero no se ha
diseminado.
✓ Retinoblastoma extraocular: se ha diseminado a los tejidos de alrededor o a otras partes del cuerpo.
✓ Retinoblastoma recurrente: el cáncer ha vuelto a aparecer o ha progresado después de haber sido tratado.
Tratamientos que contemplan la preservación de la vista
✓ Radioterapia.
✓ Crioterapia: tratamiento por congelación
✓ Fotocoagulación: se emplea una rayo láser para eliminar los vasos sanguíneos que alimentan al tumor.
✓ Termoterapia: uso de calor para destruir las células de cáncer.
✓ Quimioterapia: medicamentos que eliminan las células cancerosas; el medicamento entra en el torrente sanguíneo y puede eliminar células cancerosas en todo el cuerpo. Su utilización en niños se encuentra en estudio.
Retinoblastoma Intraocular
En un solo ojo:
✓ Si el tumor es grande y no se espera poder preservar la visión, se extrae el ojo (enucleación).
✓ Si es más pequeño y se espera preservar la visión, los métodos comentados anteriormente.
En ambos ojos
✓ Cirugía para extraer el ojo que tenga más cáncer y/o radioterapia al otro ojo.
✓ Radioterapia en ambos ojos, si existe la posibilidad de preservar la vista de ambos.
Retinoblastoma extraocular
Cualquiera de los siguientes:
✓ Radioterapia y/o Quimioterapia intratecal (espacio entre los revestimientos de la médula espinal y el cerebro).
✓ Combinaciones de medicamentos para quimioterapia y nuevas formas de administración de
medicamentos (evaluándose).
Retinoblastoma Recurrente
Depende del lugar y extensión del cáncer. Si vuelve a aparecer sólo a los ojos se realiza operación o radioterapia. Si regresa a otra parte del cuerpo, dependerá de las necesidades del paciente y otros factores.
Medidas Preventivas
Debido al factor hereditario, tanto los pacientes como sus hermanos y hermanas deben someterse a exámenes
periódicos y asesoramiento genético, para determinar el riesgo que tienen de desarrollar la enfermedad.
RETINOPATÍA DEL PREMATURO
Descripción
La retinopatía del prematuro (Fibroplastia Retrolental) (ROP) es el desarrollo anormal de los vasos sanguíneos en la retina, comienza durante los primeros días de vida y pude progresar rápidamente causando ceguera en cuestión de semanas.

Causas
Se da en algunos niños que nacen prematuramente. El aporte de sangre a la retina comienza a las 16 semanas de gestación, en el nervio óptico, y los vasos se desarrollan en ese punto hacia los bordes hasta la hora de nacer. Cuando un niño nace prematuramente este crecimiento normal se ve interrumpido y comienzan a crecer vasos anormales.
Con el tiempo, este crecimiento de los vasos produce una cicatriz de tejido fibroso que se adhiere a la retina y a la masa transparente que llena el espacio entre la retina y la cara posterior del cristalino. Se forma un anillo que puede extenderse 360 grados alrededor en el interior del ojo, puede desprender la retina y en algunos casos producir ceguera.
Factores responsables
✓ Hay una variedad de factores que pueden ser responsables del desarrollo de la retinopatía del prematuro:
*Peso bajo al nacer y la edad de gestación. Son los factores predictivos más potentes
*Incidencia de ROP en pretérminos con peso igual o menos de 1500 gr.: 26´2%.
✓ Pretérminos con peso inferior a 1000 gr.: 66%.
✓ Ventilación mecánica y la administración total de oxígeno.
✓ Niveles elevados de anhídrido carbónico en la sangre.
✓ Anemia (por cursar con hipoxia o falta de oxígeno). Tanto el aumento como la disminución de oxígeno está en relación con el ROP.
✓ Transfusiones de sangre. La hemoglobina fetal tiene mayor afinidad por el oxígeno que la del adulto por lo que las transfusiones con sangre o concentrados de hematíes procedentes de donantes adultos hacen aumentar el oxígeno libre circulante en la sangre.
✓ Hemorragias intraventriculares.
✓ Síndrome de tensión respiratoria.
✓ Hipoxia (falta de oxígeno) crónica en el útero.
✓ Convulsiones.
✓ Hay algunos autores que consideran que la exposición a la luz fluorescente de los hospitales
contribuye al desarrollo de una retinopatía del prematuro, pero hasta la fecha no ha sido
demostrado. Actualmente se piensa que el desarrollo de la enfermedad se debe a la combinación de varios factores, unos dentro del útero y otros después del nacimiento.
Epidemiología
La epidemiología es la ciencia que estudia los patrones de la enfermedad a nivel de incidencia, variaciones geográficas, demográficas, estado socioeconómico, genética, edad y causas infecciosas. Los epidemiólogos estudian la relación entre estos factores así como los patrones de migración y contribuyen a un mayor conocimiento de la enfermedad.
Síntomas
Existe una clasificación internacional que tiene en cuenta tres criterios:
✓ Situación.
✓ Extensión.
✓ Estadío:
1: demarcación clara entre retina vascular y avascular.
2: Tejido blanquecino rosado, ligeramente sobreelevado.
3: Tejido neovascular que infiltra el vítreo.
4: Desprendimiento parcial de la retina.
5: Desprendimiento total de la retina.
Hay un signo de severidad que puede darse en cualquier estadío y que indica severidad del proceso (tortuosidad vascular en el polo posterior), se denomina “plus” (+). La mayor parte de los casos con estadío 1 y 2 regresan espontáneamente, pero cuando se alcanza un estadio 3 + se calcula que el 50% de los casos evolucionarán a 4 y 5: estadío umbral en el que está indicado realizar tratamiento para detener el proceso.
Dependiendo de la etapa del ROP, el niño puede tener desde vista casi normal hasta ceguera. Muchos ROP no progresan hasta la etapa 5.
Diagnóstico
Niños a explorar:
✓ Peso de nacimiento inferior a 1600 gr.; por encima de este peso ningún niño desarrolla un ROP superior al estadío 2, y todos regresan sin experimentar cambios cicatriciales.
✓ Peso superior a 1600 gr. que han recibido oxigenoterapia prolongada o han sido sometidos a transfusiones o han padecido anemia, hipoglucemias (bajadas de azúcar) o infección importante.
La primera exploración se debe realizar a las 4 – 6 semanas de vida.
3 Pruebas
Se debe practicar una dilatación de la pupila (midriasis) y utilizar oftalmoscopio indirecto. Sin embargo, siendo el ROP el principal responsable de ceguera infantil, sería importante que se realizara una exploración completa a todos los niños prematuros.
Pronóstico
Con los avances de la neonatología ha aumentado la supervivencia de los niños prematuros de 1250 gr. o menos, y estos niños tienen el riesgo de padecer retinopatía del prematuro (ROP). En la mayoría de los casos se resuelve espontáneamente y un número pequeño progresa a estadíos severos que requieren tratamiento y algunos casos sin tratamiento o incluso con él, pueden llegar a ceguera.
Hasta hace unos años era una enfermedad considerada como una de las causas de ceguera en los niños, con nulas posibilidades de tratamiento. Hoy en día se puede reducir el riesgo de desprendimiento de retina con el tratamiento adecuado y sobre todo, con el diagnóstico precoz mediante el examen de fondo de ojo por un oftalmólogo experto en la exploración de retinas de niños prematuros dentro de las cuatro a seis semanas de nacimiento. La aparición de una clasificación internacional ampliamente aceptada y la realización de estudios multicéntricos ha permitido mejorar notablemente el manejo de la enfermedad. El conocimiento de los factores de riesgo que influyen en la aparición de la enfermedad ha contribuido a seleccionar aquellos niños pretérmino que requieren un seguimiento más estricto y buscar alternativas.
Tratamiento
El tratamiento para el ROP depende del nivel de la condición. Los niveles 1 y 2 no requieren, normalmente, más que observación.
Hay variedad de tratamientos:
Crioterapia
Se coloca un terminal muy frío fuera de la pared del ojo y congelando hasta que se forma una bola de hielo en la superficie de la retina. La ventaja principal reside en que es muy fácil tratar la zona más periférica pero es difícil acceder a zonas posteriores. Etapa 3 del ROP.
 Resultados
Resultados
✓ Aumenta el porcentaje de casos con buena visión en un 33%.
✓ Disminuye el porcentaje de casos con mala visión en un 38%
Tratamiento con láser
La fotocoagulación con láser se realiza para eliminar los vasos anormales antes que causen el desprendimiento de la retina. El haz de láser alcanza la retina avascular a través del orificio pupilar, por lo que la lesión de la esclera y tejidos circundantes es mínima. Es el tratamiento habitual. Los efectos indeseables son menores que con crioterapia. Etapa 3 del ROP.
4 Resultados
La eficacia del resultado es similar a la de la crioterapia y además disminuye los efectos indeseables del postoperatorio (lesiones en la conjuntiva, hemorragias) y a largo plazo como el riesgo de desprendimiento tardío de retina.
Vitrectomía
Se realizan pequeñas incisiones en el ojo para retirar el vítreo (sustancia transparente que ocupa la mayor parte del globo ocular, contenida en una membrana entre el cristalino y la retina) y reponerlo con una solución salina para mantener la forma y la presión del ojo. Después que el vítreo se ha retirado, el tejido cicatrizante en la retina se puede cortar, permitiendo a la retina relajarse y que vuelva a yacer sobre la pared del ojo. En la etapa 4 y 5 del ROP.
Complicaciones
Existen varias complicaciones relacionadas con el ROP que pueden presentarse después:
✓ Estrabismo (ojos cruzados).
✓ Ambliopía (oscurecimiento de la visión por sensibilidad imperfecta de la retina y sin lesión orgánica del ojo).
✓ Miopía.
✓ Glaucoma.
Es necesario hacer exámenes médicos regulares para revisar y tratar estos trastornos.
Medidas preventivas
Diagnóstico precoz mediante el examen de fondo de ojo por un oftalmólogo experto en la exploración de retinas de niños prematuros, dentro de las cuatro a seis semanas de nacimiento.
TRAUCOMA
Definición
Se trata de una infección del ojo producida por una bacteria que, si no es tratada adecuadamente, puede producir cicatrización crónica y ceguera.

Causas
La bacteria causante de la infección es la Chlamydia Trachomatis. Tras un periodo de incubación de 5 a 12 días, comienza lentamente como una conjuntivitis. Se adquiere por contacto directo con secreciones oculares o de la nariz – garganta de personas enfermas y objetos como toallas o ropa que pueden haber estado en contacto con estas secreciones. También ciertas moscas que se han alimentado de estas secreciones, pueden transmitir la enfermedad.
Epidemiología
Se presenta en todo el mundo, fundamentalmente en zonas rurales de países en desarrollo y en la edad infantil.
Sobre todo en poblaciones pobres, condiciones de vida de hacinamiento y/o mala higiene. Se considera la enfermedad de la miseria. Afecta a 150 millones de personas en todo el mundo y es la segunda causa de ceguera y la primera prevenible.
Síntomas foto
 ✓ Conjuntivitis.
✓ Conjuntivitis.
✓ Secreción ocular.
✓ Inflamación de los párpados.
✓ Inversión de las pestañas.
✓ Inflamación de los ganglios situados tras los oídos.
✓ Opacidad de la córnea. Disminuye la visión y se considera lesión invalidante.
Diagnóstico
El mejor diagnóstico es el clínico, al darse en poblaciones endémicas y a la existencia de signos suficientemente claros. El diagnóstico definitivo se realiza mediante la detección del organismo o el antígeno en raspados en la conjuntiva o por aislamiento de la bacteria en cultivo.
Pronóstico
Mediante el tratamiento oportuno, el pronóstico es muy bueno. Si no es así:
✓ Cicatrización de la conjuntiva y la córnea.
✓ Deformaciones de los párpados.
✓ Inversión de las pestañas.
✓ Pérdida de la visión.
Tratamiento
Antibióticos por vía oral.
La Organización Mundial de la Salud ha desarrollado un programa simplificado de diagnóstico, prevención y tratamiento. El tratamiento médico convencional tan sólo disminuye la gravedad de la enfermedad durante uno o dos meses si no se suministra agua a las poblaciones afectas. Cuando nos encontremos frente a estadios activos de la enfermedad, los trataremos médicamente. En el caso de que nos encontremos frente a estadios cicatriciales es necesario un tratamiento quirúrgico.
 Tratamiento médico
Tratamiento médico
En poblaciones endémicas, el tratamiento individualizado no sirve para nada. El tratamiento masivo consiste en administrar de forma tópica (pomada) a todos los miembros de la comunidad:
✓ Tetraciclina 1% cada 12 horas, durante 16 semanas.
✓ Tetraciclina 1% cada 12 horas, 5 días al mes, durante 6 meses.
✓ Tetraciclina 1% cada 24 horas, 10 días al mes, durante 6 meses.
El tratamiento sistémico selectivo:
✓ Tetraciclina oral, 250 mg cada 6 horas, durante 3 semanas. ( en niños mayores de 7 años)
✓ Doxiciclina, 100 mg cada 24 horas, durante 6 semanas. (en niños mayores de 7 años)
✓ Eritromicina, 250 mg cada 6 horas, durante 3 semanas. (en recién nacidos y embarazadas)
✓ Cotrimoxazol, 2 comprimidos cada 48 horas, durante 3 semanas.
✓ La azytromicina: una dosis única por vía oral podría ser tan efectiva como seis meses de tratamiento intermitente con tetraciclina
Tratamiento quirúrgico
Existen multitud de técnicas y, dado que la mayoría son realizadas por OMAS (ophthalmic medical assistants) - pues hay zonas con una dramática carencia de oftalmólogos (1/6.000.000)-, ha sido imposible realizar estudios que demuestren las ventajas de unas sobre otras.
Medidas Preventivas
La disponibilidad de agua, la higiene y el cambio de determinados hábitos pueden disminuir la incidencia en un 50%.
Hay estudios para el desarrollo de una vacuna, pero las características de la bacteria dificultan la posibilidad de obtener resultados a corto plazo.
SÍNDROME DE WOLFRAM
SÍNDROME DE WOLFRAM
Descripción
El síndrome de Wolfram es una enfermedad neurodegenerativa también conocida por el término acrónimo DIDMOAD (DIDMAOS en español), que se refiere a sus principales componentes clínicos: Diabetes Insípida, Diabetes Mellitus, Atrofia Óptica y Sordera.
Etiopatogenia
Las causas de este síndrome no se conocen con exactitud todavía. En la mayoría de los casos se evidencia una herencia autosomica recesiva, (es decir que se tienen que heredar la versión alterada del gen de ambos progenitores para padecer la enfermedad), aunque en algunos pacientes parece existir cierta herencia mitocondrial, (que seria solamente de herencia materna).El síndrome de Wolfram es una enfermedad genéticamente muy heterogénea, se da con la misma frecuencia en hombres y mujeres; y por ahora no se han encontrado casos en razas no caucásicas. El gen de la wolframina, cuyas mutaciones podrían ser parte de la base de la enfermedad, ha sido mapeado en el cromosoma 4p16.1. La wolframina se encuentra en células de todo el organismo, siendo sus concentraciones más altas en el corazón, cerebro, pulmones y páncreas; aunque su función se desconoce por ahora parece jugar un papel en el procesamiento de otras proteínas y en la supervivencia de las células nerviosas y pancreáticas.
Manifestaciones clínicas
Se trata de un síndrome con enormes variedades clínicas, la severidad de cada uno de los componentes que lo integran varia de paciente a paciente, al igual que la velocidad de aparición; pacientes incluso de la misma familia pueden padecer el síndrome con una severidad distinta. Los componentes esenciales son la Diabetes Mellitus y la Atrofia Óptica.

Normalmente la diabetes mellitus es el primer componente en aparecer y suele hacerlo en la primera década junto con la atrofia del nervio óptico que aparece después. La perdida progresiva y bilateral de las fibras del nervio óptico hacen que la visión se vaya deteriorando de manera progresiva, lo que en la mayoría de los casos, alrededor de la tercera década, desemboca en la ceguera.
La diabetes insípida central aparece generalmente en la segunda década, se trata de un trastorno en la que por el déficit de la hormona vasopresina, el organismo es incapaz de concentrar la orina; esto hace que se eliminen grandes cantidades de orina muy diluida que pueden llevar a la deshidratación del paciente.
La sordera sensorial, bilateral y simétrica es otro componente característico del síndrome. Suele aparecer entre la primera y segunda década y aunque al principio afecta mas a las frecuencias agudas, su curso progresivo hace que en la mayoría la audición se vea afectada de manera intensa
Los pacientes suelen además presentar alteraciones del aparato urinario, atrofia gonadal y múltiples síntomas neurológicos acompañados de alteraciones psiquiatricas de tipo alteraciones conductuales y conductas suicidas. La muerte ocurre de manera temprana, muchas veces debido al fallo respiratorio ocasionado por la degeneración progresiva del tronco del encéfalo.
Diagnóstico
Debido a la complejidad genética del síndrome de Wolfram el diagnostico hoy por hoy sigue siendo clínico. Es importante la vigilancia en los jóvenes con diabetes mellitus de la aparición de cualquiera de los otros síntomas del DIDMOAD. LA aparición es generalmente secuencial aunque no siempre sigue un orden concreto. Es importante el diagnostico precoz debido a la naturaleza progresiva de la enfermedad lo que hace que el control de las complicaciones a largo plazo pueda mejorar el pronostico de la misma. También es muy importante el consejo genético debido a su componente hereditario.
El diagnóstico diferencial debe hacerse con otras enfermedades que asocien atrofia óptica y sordera hereditaria:
- El síndrome de Alström (diabetes mellitus juvenil, sordera, atrofia óptica debido a la degeneración retiniana, obesidad e hipogonadismo).
- El síndrome de Refsum (diabetes mellitus juvenil, sordera, retinitis pigmentaria atípica, polineuritis, ictiosis, lesiones cardíacas y depósito de ácido fitánico).
- Del síndrome de Bardet-Biedl (retinitis pigmentaria atípica, obesidad, hipogonadismo, polidactalia e inteligencia reducida).
- La enfermedad de Schuller-Christan (exoftalmos, atrofia óptica, defectos craneales y diabetes mellitus).
- Debería excluirse el craneofaringioma, el tumor más común que causa la atrofia óptica y la diabetes insípida.
Tratamiento
No existe hoy en día ningún tratamiento curativo de la enfermedad. El objetivo del tratamiento es el control de los síntomas y evitar los efectos deletéreos de la diabetes mellitus y de la diabetes insípida, lo que en muchas ocasiones, como ya se dijo condiciona el pronostico. Por la diversidad y gravedad de las anomalías que presentan es fácil entender que estos pacientes deben ser diagnosticados y seguidos por diferentes especialistas, pero en el marco de un equipo multidisciplinar común que coordine y oriente las actitudes a tomar. Este enfoque se viene realizando con éxito en varios hospitales de Europa y Estados Unidos. En España sin embargo no existe por ahora ninguna unidad de este tipo, aun cuando existe una demanda real tanto por parte de los pacientes afectados como por los propios facultativos.
El tratamiento se dirigirá al control de la diabetes mellitus con dieta asociada o no a fármacos; y al control de la diabetes insípida mediante la administración de vasopresina o clorpropamida para la producción de hormona antidiurética. La pérdida auditiva sensorial puede reducirse con la utilización de ayuda auditiva. Se han publicado casos de recuperación visual sólo en algunas ocasiones mediante la destrucción quirúrgica de las adherencias de la aracnoiditis optoquiasmática. La cirugía puede ser necesaria en casos de uropatía.












